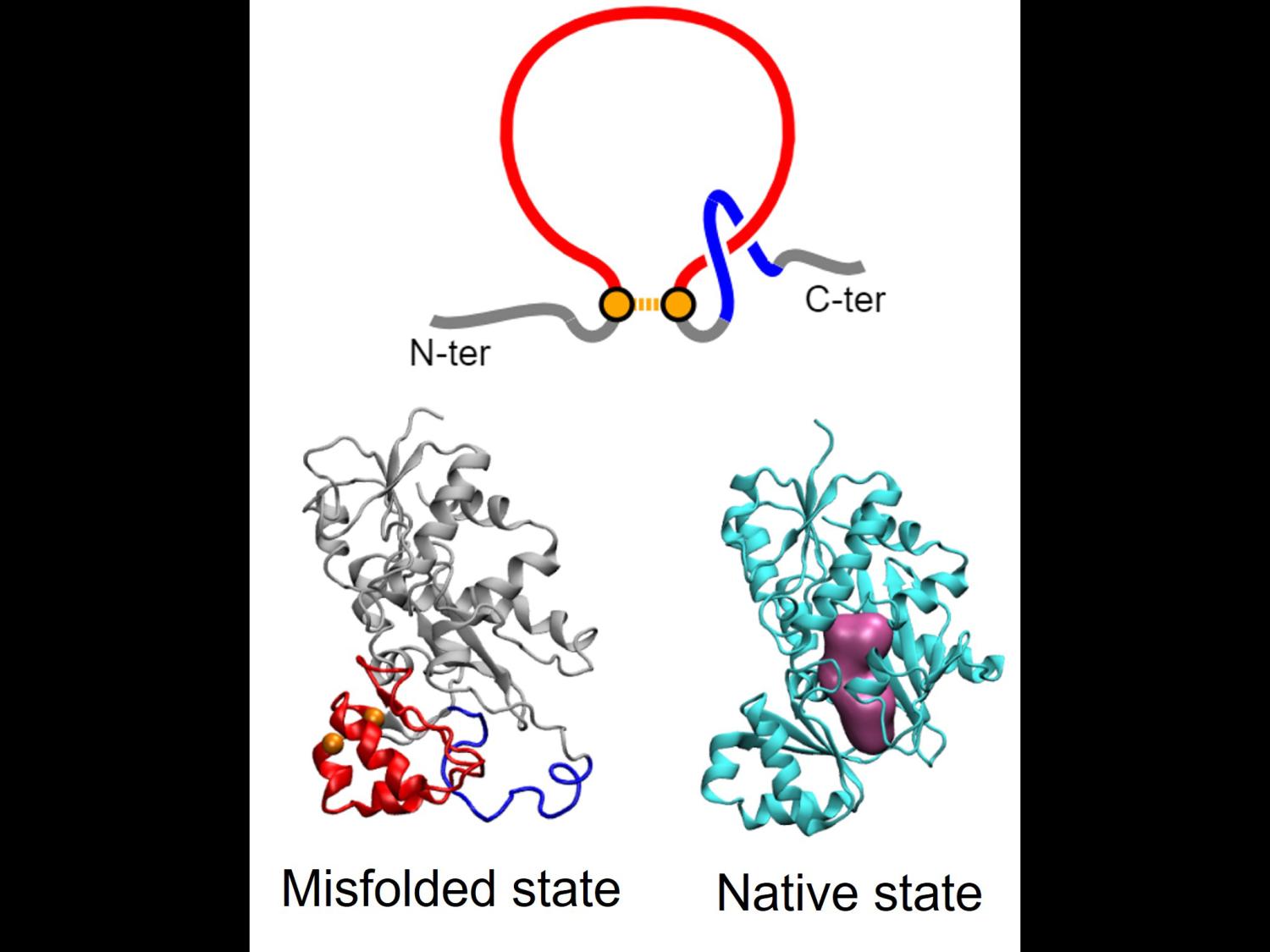
New modeling shows how synonymous mutations that change the DNA sequence of a gene, but not the sequence of the encoded protein can impact protein production and function by changing the rate of protein synthesis. Top: illustration of a new class of protein misfolding called a non-covalent lasso entanglement that can result from changes to the rate of protein synthesis caused by synonymous mutations. Bottom: structure of a protein showing its native state and misfolded state with non-covalent lasso entanglement. Credit: Yang Jiang, Penn State.
Why synonymous mutations are not always silent
Posted on December 6, 2022UNIVERSITY PARK, Pa. — New modeling shows how synonymous mutations — those that change the DNA sequence of a gene but not the sequence of the encoded protein — can still impact protein production and function. A team of researchers led by Penn State chemists modeled how genetic changes that alter the speed of protein synthesis, but not the sequence of amino acids that comprise the protein, can lead to misfolding that changes the protein’s activity level, and then corroborated their models experimentally. The results demonstrate the importance of kinetics — the rate of protein synthesis — in addition to sequence for determining protein structure and function and could have implications in fields such as biopharmaceutics for fine tuning the activity of synthesized proteins.
Proteins are composed of long strings of amino acids that then fold up into three-dimensional functional structures. Each amino acid is encoded by a triplet of letters in the DNA alphabet of A, T, C and G called a codon, but there is redundancy built in to the system such that more than one codon can correspond to the same amino acid. Therefore, a mutation that changes the DNA sequence of a gene won’t necessarily change the sequence of the encoded protein if the mutation results in a “synonymous codon.” To make a protein, DNA in the nucleus of a cell is first transcribed into a messenger RNA (mRNA). The mRNA is then transported out of the nucleus where it is translated into a nascent protein by a cellular organelle called a ribosome. After translation the protein is folded into its final functional form.
“We used to use ‘synonymous’ and ‘silent’ interchangeably to describe mutations that don’t change a protein’s sequence because it was thought that they wouldn’t alter the function of the protein,” said Ed O’Brien, professor of chemistry and a co-hire of the Institute for Computational and Data Sciences at Penn State, and one of the leaders of the research team. “But, we’ve known for some time now that not all synonymous mutations are silent. Over two decades ago, it was shown that synonymous mutations could reduce the activity of proteins, but it was still unknown what was happening at the molecular level to cause this change.”
The research team used a multi-scale modeling approach, using theory and computation to simulate what is happening at the molecular level during protein synthesis, to predict changes in protein structure that could result from synonymous mutations and therefore alter the protein’s activity. A paper describing the research appears Dec. 5 in the journal Nature Chemistry.
“For a variety of reasons, some codons are translated at different speeds by the ribosome,” said Yang Jiang, assistant research professor of chemistry at Penn State and the first author of the paper. “For three different enzymes — specialized proteins that catalyze biochemical reactions — we simulated one version of the mRNA composed of fast translating codons and one version composed of slowly translating codons and then modeled the production of the nascent protein, how it is folded post-translationally, and its activity.”
The team’s predictions for changes in protein activity matched experimental results that had been measured previously for one of the enzymes. Experiments were then performed for the other two enzymes that also matched the changes in activity predicted by their modeling. They then examined the predicted protein structures and folding pathways from their models to try to identify changes at the molecular level that could have led to the changes in activity.
“In our models we found a new class of protein misfolding that we call a ‘non-covalent lasso entanglement,’” said Jiang. “Essentially, a portion of the protein forms a closed loop and one end of the protein incorrectly threads through the loop and gets trapped for long time periods.”
The researchers suggest two potential reasons that this form of misfolding can reduce the protein’s activity. First, the misfolding occurs near the active site of the enzymes, which can disrupt its activity. Second, while cells have mechanisms called chaperones that can refold or remove misfolded proteins, these particular misfolded structures may be subtle enough to not be recognized by the chaperone system and they can persist in the cell because the observed changes would require a large portion of protein to be unfolded in order to correct them.
“So, the question then is ‘How is this happening?’ and we can use our models to follow the folding pathway of the protein to address this,” said O’Brien. “We see inflection points during folding where the protein can either travel down a path that leads to a correctly folded protein or it can take a path that leads to the lasso entanglement. We call this ‘kinetic partitioning.’ How fast or slowly the protein is being translated — the kinetics of the process — seems to influence which path the protein is more likely to take.”
These new insights into how the kinetics of protein synthesis can influence protein structure and function could have repercussions in fields ranging from biochemistry to biotechnology and to medicine.
“The predominant paradigm in the field of protein folding has been that the sequence determines structure,” said O’Brien. “Our results provide an explanation and illustration of how kinetics can also control protein structure and function. This has implications for any field involving protein synthesis. Protein misfolding also contributes to some human diseases, so our work indicates an entirely new class of drug targets may exist for the development of future drugs.”
In addition to O’Brien and Jiang, the team includes Syam Sundar Neti, Ian Sitarik, Priya Pradhan, and Squire J. Booker at Penn State; and Philip To, Yingzi Xia, and Stephen D. Fried at Johns Hopkins University. The research was funded by the U.S. National Institutes of Health and the U.S. National Science Foundation. Additional support was provided by the Howard Hughes Medical Institute and the Penn State Institute for Computational and Data Sciences.